The secret of life, part 2: the solution of the protein folding problem.
20 years ago, the sequencing of the human genome gave us our own blueprint, right? Wrong — it actually gave us an encrypted blueprint! Every gene is there, but information about its function is effectively encrypted. Today’s announcement at #CASP14 is a universal decryption key, which scientists have been hunting for 50 years, that will remove a bottleneck to mechanistic modelling and understanding across all biology.
Biology is hierarchical: The genome specifies how to make RNA transcripts which are translated to make proteins; proteins are the machines that compose and build cells; cells together form organs which make up a whole organism. In theory a complete genome contains all the information to specific a model of a complete organism, provided the rules to build each layer of hierarchy are understood. The rules of translation — how 4 letters of DNA encode the 20 types of amino acids that make up proteins — both linear molecules — were decoded by 1966. However, the sequence of a protein encodes how it spontaneously folds up in 3D, with the resulting shape determining its function. Until today we hadn’t been able to decode the rules of this folding process — at least not well enough to infer the next layer of the biological system systematically, the complete set of protein structures. At this level the genome has remained the equivalent of an encrypted disk drive without the decryption key.
Researchers have been able to partially work around this: protein 3D structures can be determined by a variety of experimental methods: Xray, NMR and most recently cryo-electron microscopy. However, experimental processes are hard and slow, so the set of protein structures is far from a complete for any organism, e.g. for human only about 17% of the protein sequence encoded by the genome has an experimental 3D structure despite decades of effort.
A schematic of what this has meant for biological research is shown in figure 1 below. The absence of a complete set of 3D protein structures has limited our ability to project information from the genome upwards to directly infer higher organisational structures of biology. Instead, advances in understanding have depended on experimental collection of intermediates: transcripts, epigenetic states, structures, cells etc. and the experimental investigation of different components by tens of thousands of researchers worldwide.
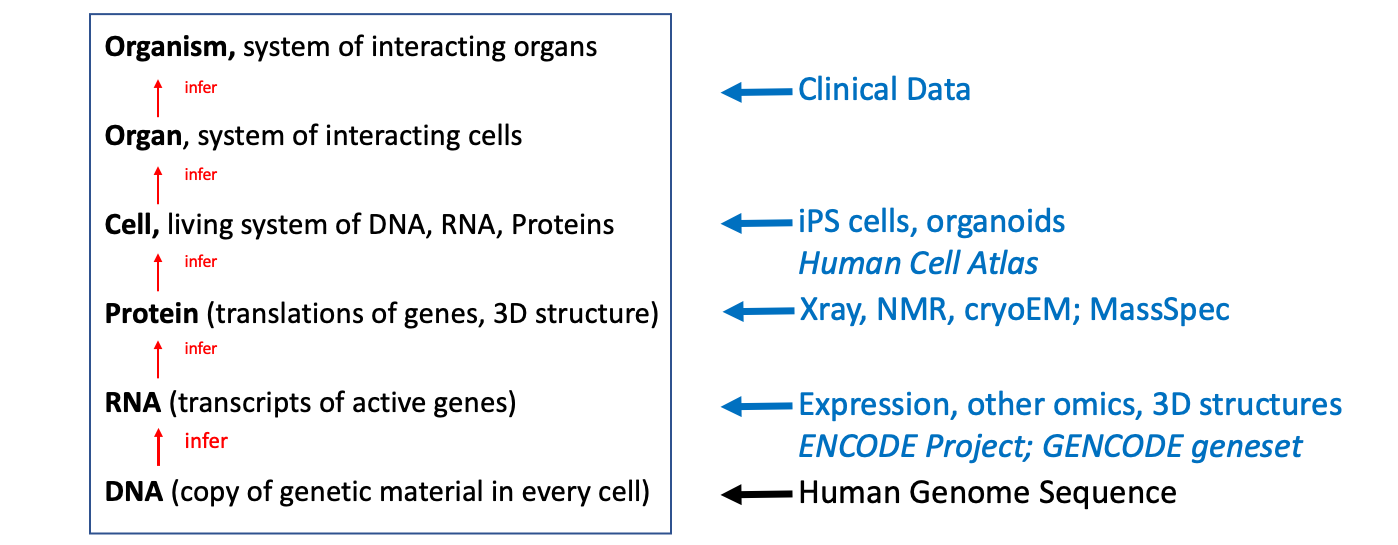
Hence the 50 year worldwide hunt for a method to decode protein folding, the last 26 years of which has been carried out under the auspices of bi-annual blind test evaluation of methods, CASP. The announcement today, that Deepmind’s Alphafold artificial intelligence (AI) algorithm can predict most protein structures to experimental accuracy, represents an amazing breakthrough and brings the prospect of complete sets of protein 3D structures being rapidly generated for all organisms, including human.
A schematic of what the solution to the folding problem can mean is shown in figure 2 below. With a complete set of protein 3D structures, it becomes practical to build complete mechanistic models of biological processes like transcription, regulation and progressively infer higher organisational structures of biology directly, relying less on of generation of intermediate datasets. Mapping variants onto more complete mechanistic models will also enhance our ability to infer the consequence of sequence differences in the genome, with implications for personalised healthcare.

Beyond these implications there are many other aspects of the scientific breakthrough announced today by Deepmind and CASP. It’s a lesson in the benefits of structures to support team science with engagement across an entire world community and embedded evaluation and openness of results. The development of the successful approach builds on the progressive developments and progress exposed through 26 years of the CASP process. It’s also a demonstration of the power of AI methods to model systems where arguably classical physics approaches fail, with implications for many other problems. It’s also arguably the first case where AI really will change the world and in a positive way — having an impact far beyond its ability to win games of Chess or Go, however impressive those achievements are.
There are still gaps in the current AI solution: extending from predicting structures of isolated protein monomers to multimers; extending from predicting structures to predicting interactions between structures. However there is huge activity and progress, and while Deepmind are well ahead in CASP14, it’s clear from progress by other groups using AI techniques developed since CASP13 that there isn’t going to be a monopoly in algorithm development in this space, which will also help drive further refinement and extension. Collectively these applications of AI will progressively transform our ability to model biological systems, similar to the sweeping impact of the wide availability of genome sequences and genome sequencing.
Tim Hubbard is Professor of Bioinformatics at King’s College London, with roles at Genomics England and Health Data Research UK. He was a co-organiser of CASP 1996–2007.
